





Kromě technologie byla syntéza glykosidů vždy zajímavá i pro vědu, protože se jedná o velmi běžnou reakci v přírodě. Nedávné práce Schmidta a Toshimy a Tatsuty, stejně jako mnoho citovaných odkazů, se zabývají širokou škálou syntetických možností.
Při syntéze glykosidů se vícecukerné složky kombinují s nukleofily, jako jsou alkoholy, sacharidy nebo proteiny. Pokud je vyžadována selektivní reakce s jednou z hydroxylových skupin sacharidu, musí být v prvním kroku chráněny všechny ostatní funkce. V principu mohou enzymatické nebo mikrobiální procesy díky své selektivitě nahradit složité chemické kroky ochrany a deprotekce a selektivně oddělit glykosidy v určitých oblastech. Vzhledem k dlouhé historii alkylglykosidů však nebylo použití enzymů při syntéze glykosidů dosud široce studováno a aplikováno.
Vzhledem k kapacitě vhodných enzymatických systémů a vysokým výrobním nákladům není enzymatická syntéza alkylpolyglykosidů připravena k povýšení na průmyslovou úroveň a upřednostňují se chemické metody.
V roce 1870 M.A. Colley popsal syntézu „acetochlorhydrózy“ (1, obrázek 2) reakcí dextrózy (glukózy) s acetylchloridem, což nakonec vedlo k historii syntézních cest glykosidů.
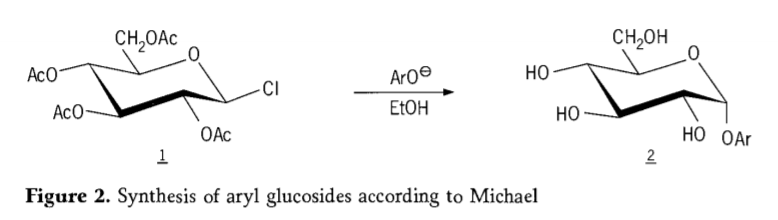
Tetra-0-acetyl-glukopyranosylhalogenidy (acetohaloglukózy) se později ukázaly jako užitečné meziprodukty pro stereoselektivní syntézu čistých alkylglukosidů. V roce 1879 se Arthurovi Michaelovi podařilo připravit definitivní, krystalizovatelné arylglykosidy z Colleyho meziproduktů a fenolátů (Aro-, obrázek 2).
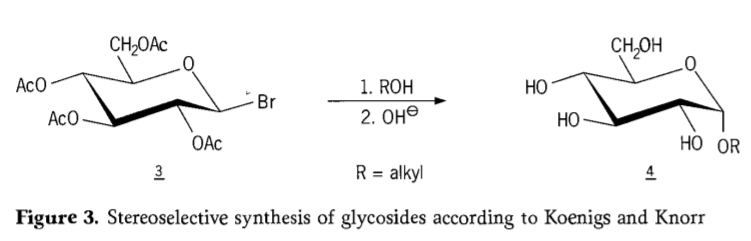
V roce 1901, kdy W. Koenigs a E. Knorr představili svůj vylepšený stereoselektivní glykosidační proces (obrázek 3), Michaelova syntéza široké škály sacharidů a hydroxylových aglykonů. Reakce zahrnuje substituci SN2 na anomerním uhlíku a probíhá stereoselektivně s inverzí konfigurace, čímž vzniká například α-glukosid 4 z β-anomeru aceobromglukózového meziproduktu 3. Koenigs-Knorrova syntéza probíhá v přítomnosti stříbrných nebo rtuťových promotorů.
V roce 1893 navrhl Emil Fischer zásadně odlišný přístup k syntéze alkylglukosidů. Tento proces je dnes dobře známý jako „Fischerova glykosidace“ a zahrnuje kysele katalyzovanou reakci glykos s alkoholy. Jakýkoli historický záznam by však měl zahrnovat i první zaznamenaný pokus A. Gautiera z roku 1874 o přeměnu dextrózy bezvodým ethanolem za přítomnosti kyseliny chlorovodíkové. Kvůli zavádějící elementární analýze se Gautier domníval, že získal „diglukózu“. Fischer později prokázal, že Gautierova „diglukóza“ byla ve skutečnosti převážně ethylglukosid (obrázek 4).
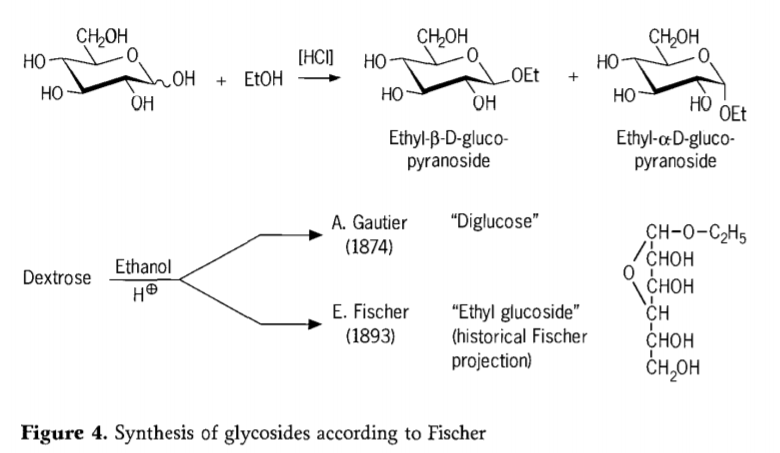
Fischer správně definoval strukturu ethylglukosidu, jak je patrné z navrženého historického furanosidového vzorce. Fischerovy glykosidační produkty jsou ve skutečnosti komplexní, většinou rovnovážné směsi α/β-anomerů a izomerů pyranosidů/furanosidů, které také obsahují náhodně vázané glykosidové oligomery.
V důsledku toho není snadné izolovat jednotlivé molekulární druhy z Fischerových reakčních směsí, což byl v minulosti vážný problém. Po určitém vylepšení této syntetické metody Fischer následně přijal pro svá zkoumání Koenigsovu-Knorrovu syntézu. Pomocí tohoto postupu E. Fischer a B. Helferich jako první v roce 1911 ohlásili syntézu alkylglukosidu s dlouhým řetězcem, který vykazuje povrchově aktivní vlastnosti.
Již v roce 1893 si Fischer správně všiml základních vlastností alkylglykosidů, jako je jejich vysoká stabilita vůči oxidaci a hydrolýze, zejména v silně alkalickém prostředí. Obě vlastnosti jsou cenné pro alkylpolyglykosidy v aplikacích povrchově aktivních látek.
Výzkum týkající se glykosidační reakce stále probíhá a v nedávné minulosti bylo vyvinuto několik zajímavých cest ke glykosidům. Některé postupy syntézy glykosidů jsou shrnuty na obrázku 5.
Obecně lze chemické glykosidační procesy rozdělit na procesy vedoucí ke komplexním oligomerním rovnováhám v kyselinou katalyzované glykosylové výměně.
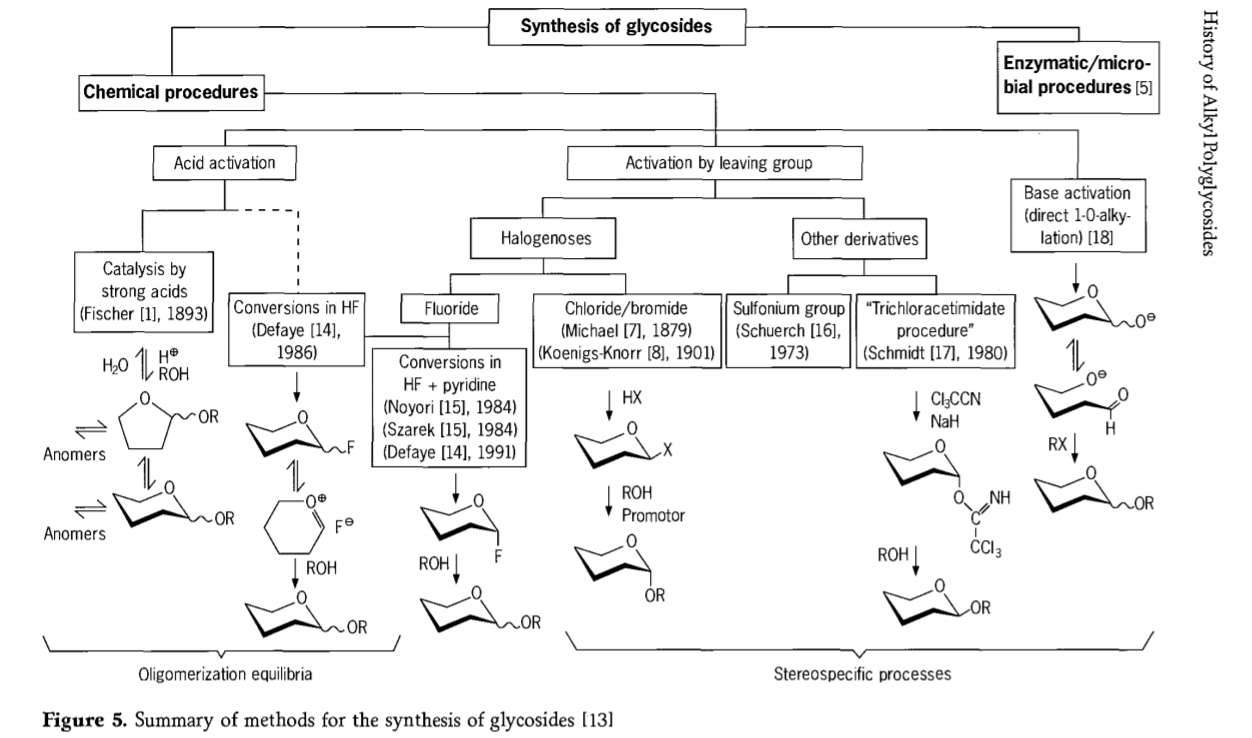
Reakce na vhodně aktivovaných sacharidových substrátech (Fischerovy glykosidické reakce a reakce fluorovodíku (HF) s nechráněnými molekulami sacharidů) a kineticky řízené, nevratné a převážně stereotaktické substituční reakce. Druhý typ postupu může vést k tvorbě jednotlivých druhů spíše než ke komplexním směsím reakcí, zejména v kombinaci s technikami konzervačních skupin. Sacharidy mohou zanechávat skupiny na ektopickém uhlíku, jako jsou atomy halogenů, sulfonyly nebo trichloracetimidátové skupiny, nebo mohou být aktivovány bázemi před přeměnou na triflátové estery.
V konkrétním případě glykosidací ve fluorovodíku nebo ve směsích fluorovodíku a pyridinu (pyridinium poly[fluorovodík]) se glykosylfluoridy tvoří in situ a plynule se přeměňují na glykosidy, například s alkoholy. Fluorovodík se ukázal jako silně aktivační, nedegradující reakční médium; rovnovážná autokondenzace (oligomerizace) je pozorována podobně jako u Fischerovy reakce, ačkoli reakční mechanismus je pravděpodobně odlišný.
Chemicky čisté alkylglykosidy jsou vhodné pouze pro velmi speciální aplikace. Například alkylglykosidy byly úspěšně použity v biochemickém výzkumu pro krystalizaci membránových proteinů, jako je trojrozměrná krystalizace porinu a bakteriorodopsinu za přítomnosti oktyl-β-D-glukopyranosidu (další experimenty založené na této práci vedly k Nobelově ceně za chemii pro Deisenhofera, Hubera a Michela v roce 1988).
Během vývoje alkylpolyglykosidů byly v laboratorním měřítku používány stereoselektivní metody k syntéze řady modelových látek a ke studiu jejich fyzikálně-chemických vlastností. Vzhledem k jejich složitosti, nestabilitě meziproduktů a množství a kritické povaze odpadních produktů v procesu by syntézy typu Koenigs-Knorr a další techniky s ochrannými skupinami vytvářely značné technické a ekonomické problémy. Procesy Fischerovy metody jsou v porovnání s ostatními méně složité a snadněji se provedou v komerčním měřítku, a proto jsou preferovanou metodou pro výrobu alkylpolyglykosidů ve velkém měřítku.
Čas zveřejnění: 12. září 2020